








外用药物制剂体外透皮试验技术要求概况
外用药物制剂体外透皮试验技术要求概况
潘宪伟1,苏 梅1,刘 新1,李慧敏2*,殷连珍1
(1. 江苏柯菲平医药股份有限公司,南京;2. 南京正大天晴制药有限公司,南京)
期刊:沈阳药科大学学报; 网络首发:2023年11月23日
第一作者:潘宪伟; 通讯作者:李慧敏,殷连珍
摘要:外用药物制剂具有复杂的药物递送途径,通常局部起效,辅料成分复杂,处方组成和制备工艺参数的微小差异就有可能导致产品出现不同的质量特性,从而影响药物的安全性和有效性。近年来,体外透皮试验(in vitro permeation test,IVPT)被广泛用于外用药物制剂的开发及其生物等效性评价。但该试验操作复杂,影响因素较多,操作不当,就有可能导致不同试验之间的结果无法比较,进而影响此类制剂的安全性和有效性评价。本文参考国内外相关技术指导原则及文献,对外用药物制剂IVPT研究的技术要求进行综述,期望为业内人士提供参考。
关键词:外用药物制剂;经皮给药;体外透皮试验(IVPT);一致性评价
外用药物制剂是一类发挥局部或全身治疗作用的制剂,剂型包括软膏剂、乳膏剂、凝胶剂、散剂、乳剂、糊剂、混悬剂、喷雾剂、气雾剂、溶液和其他半固体和/或液体制剂[1, 2]。该类制剂的质量测试分为两类:一类是评估产品质量属性的测试,包括物理化学和结构特性(Q3),如外观、混悬药物的晶型、粒度分布、液滴粒径、流变特性、pH值、黏度、含量/含量均匀度、微生物限度、有关物质、抑菌剂含量、抗氧剂含量和无菌等;另一类是评估产品性能的测试,包括体外释放试验(IVRT)和IVPT[1, 3, 4]。
外用药物制剂在处方组成(Q1)和用量(Q2)一致性的基础上,对Q3特性进行评估是生物等效性豁免的重要基础[3, 5, 6]。在此基础之上,IVPT可用于表征外用药物制剂中活性成分达到作用部位的速率和程度,可通过论证IVPT的等效性,证明生物利用度的速率和程度无显著差异,用于支持生物等效性论证或代替临床治疗等效性研究[3, 7];此外,可通过IVRT和IVPT研究,对A、B、C水平的处方变更进行评估[8]。
然而,IVPT试验结果受多种因素的影响,如:扩散池的选择;样品分析方法;生物膜(皮肤)的来源、处理和保存;试验设计(如:上样量、上样技术;接收介质的种类、pH值和离子强度;设备平衡;试验时长和采样)等。上述参数的不一致,不同操作者及实验室之间均可能导致结果重现性差,试验结果不可靠。目前国内暂无相关指南对其进行统一规定;在2022年4月之前,国外亦无相关药典对其进行明确要求。本文根据美国药典(USP)2022年5月发布的“PF 48(3) <1724> Semisolid drug products-performance tests”[9],以及2022年10月美国食品药品监督管理局(FDA)发布的行业指南“In vitro permeation test studies for topical drug products submitted in ANDAs”[2],同时结合FDA、欧洲药品管理局(EMA)和日本医药食品局(PMDA)发布的其他指南及相关文献,对外用药物制剂的体外透皮试验技术要求概况进行总结,期望为业内人士提供参考。
1 国内外政策法规
近年来,我国开始逐步规范和提高皮肤外用药物制剂的审评要求。2018年、2020年和2022年国家药品监督管理局药品审评中心(CDE)分别发布《新注册分类的皮肤外用仿制药的技术评价要求(征求意见稿)》[10]、《化学仿制药透皮贴剂药学研究技术指导原则(试行)》[11]和《局部给药局部起效药物临床试验技术指导原则》[12],对皮肤局部外用制剂研发过程中的参比制剂、处方工艺开发、原辅包质量控制、质量研究与控制、稳定性研究和等效性评价方法等方面提出了明确的技术要求。但尚未发布关于IVPT研究相关的技术要求。
USP、FDA、EMA和PMDA,从1997年至今发布的外用药物制剂质量研究相关指南及概况如表1所示。
Table 1 Related Policies and Technical Regulations for Topical Products in Different Countries or Regions表1 不同国家或地区外用药物制剂的相关政策及技术法规
Table 1 Related Policies and Technical Regulations for Topical Products in Different Countries or Regions表1 不同国家或地区外用药物制剂的相关政策及技术法规
机构 | 法规名称 | 发布年份 | 法规概述 |
USP | General Chapters 3: Topical and transdermal drug products-product quality tests[4] | 2023 | 主要阐述外用和透皮制剂关键质量属性 |
PF48(3): General Chapters 1724: Semisolid drug products-performance tests[9] | 2022 | 是目前IVRT 和IVPT研究设计和设备确认较为全面的通则,但不包括IVPT统计分析和方法学验证 | |
FDA | SUPAC-SS: Nonsterile semisolid dosage forms; scale-up and postapproval changes: chemistry, manufacturing, and controls; in vitro release testing and in vivo bioequivalence documentation[13] | 1997 | 重点阐述半固体制剂放大生产和批准后变更相关研究,对IVRT的研究设计及统计分析进行了介绍,但不包括IVPT研究设计和方法验证 |
Draft guidance on acyclovir[14] | 2016 | 包括IVRT和IVPT的研究设计和验证,其中部分参数相对USP<1724>[9]较为宽松,目前FDA已删除该指南,要求IVPT按USP<1724>和FDA IVPT[2]进行研究 | |
In vitro permeation test studies for topical drug products submitted in ANDAs[2] | 2022 | 对皮肤屏障完整性测试进行阐述,并非常详细的给出IVPT方法验证、正式研究和统计分析相关要求 | |
Physicochemical and structural (Q3) characterization of topical drug products submitted in ANDAs[3] | 2022 | 阐述外用药物制剂的物理化学和结构特性(Q3),以及IVPT研究在生物等效性评估中的作用 | |
欧盟 | Guideline on quality of transdermal patches[15] | 2014 | 对IVRT和IVPT的研究设计内容,阐述较为简单,且不包括设备的相关确认,亦未阐述方法验证内容 |
Draft guideline on quality and equivalence of topical products[7] | 2018 | ||
日本 | Guideline on biological equivalence for formulation changes of topical products (semisolid products and patches)[8] | 2010 | 重点阐述皮肤局部适用制剂(半固体制剂和贴剂)处方变更生物等效性研究,对IVRT和IVPT的研究设计内容,阐述较为简单,且不包括设备的相关确认,亦未阐述方法验证内容 |
Guideline on biological equivalence for formulation changes of topical products (semisolid products and patches): questions and answers[16] | 2010 |
2 设备确认
IVPT研究中常用的扩散池主要有立式扩散池(VDC)和流通池(FDC),USP收载的3种型号VDC设计示例见图1-3、FDC设计示例见图4。无论扩散池生产商提供了何种确认信息,实验室内部都应对每个扩散池进行再确认,至少包括:①测量供给室和接收室之间膜安装孔口处的扩散面积;②测量VDC接收室的容积或FDC输出管的长度;③在整个研究期间,膜表面温度的稳定性;④如适用,VDC的搅拌速率,或FDC的流速[9]。
根据USP<1724>,对于上述确认内容,要求:①供给室和接收室的孔口直径应限定在规定范围的±5%之内;②在测定接收室体积时,应先把所有组件(如搅拌子)配置到相应位置,将测定的接收室体积精确到0.01 mL(如6.89 mL),在实际计算时不应使用标称接收室体积(如7 mL)代替准确测定的VDC体积;③一旦接收介质和皮肤的温度达到平衡(如30 min后),应测量皮肤表面温度,确保在上样之前,皮肤表面温度维持在目标温度(32 ℃)的±1 ℃范围内,不应假定皮肤表面温度与接收液温度、或循环水浴、或干热系统的设定值、或系统的任何其他组成部分的温度相同,而应直接测定皮肤表面温度(使用红外温度计或热电偶);④VDC搅拌速率通常在400~600 rpm范围内;⑤应根据包括设备使用范围、实验室的风险承受能力和制造商建议在内的因素在6~12个月进行再认证。
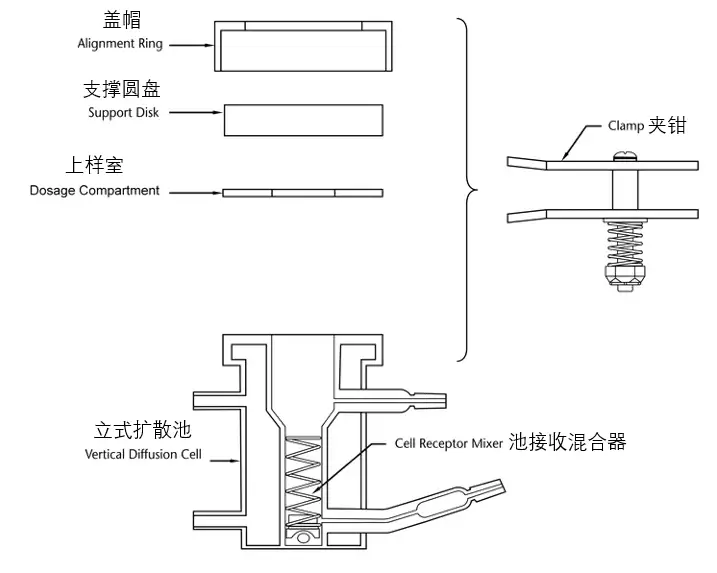
Fig.1 Example of Possible Design for VDC — Model A图1 VDC(模型A)可能采用的设计示例
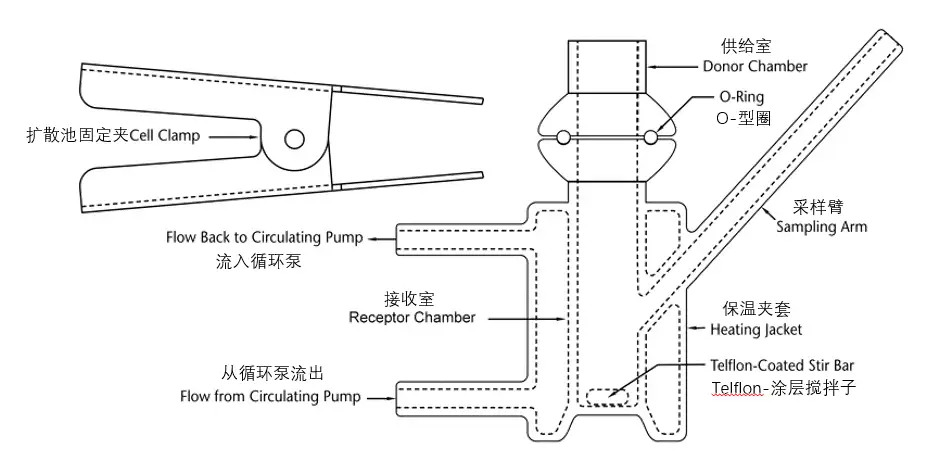
Fig.2 Example of Possible Design for VDC — Model B图2 VDC(模型B)可能采用的设计示例
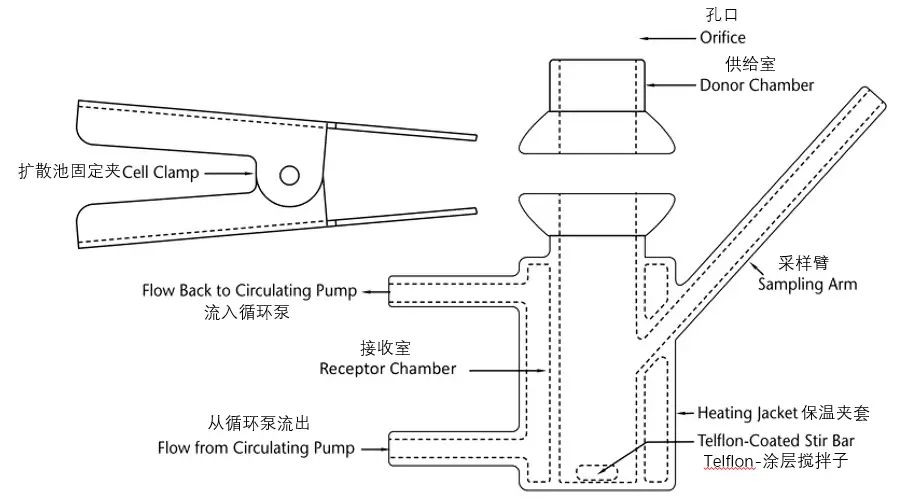
Fig.3 Example of Possible Design for VDC — Model C图3 VDC(模型C)可能采用的设计示例
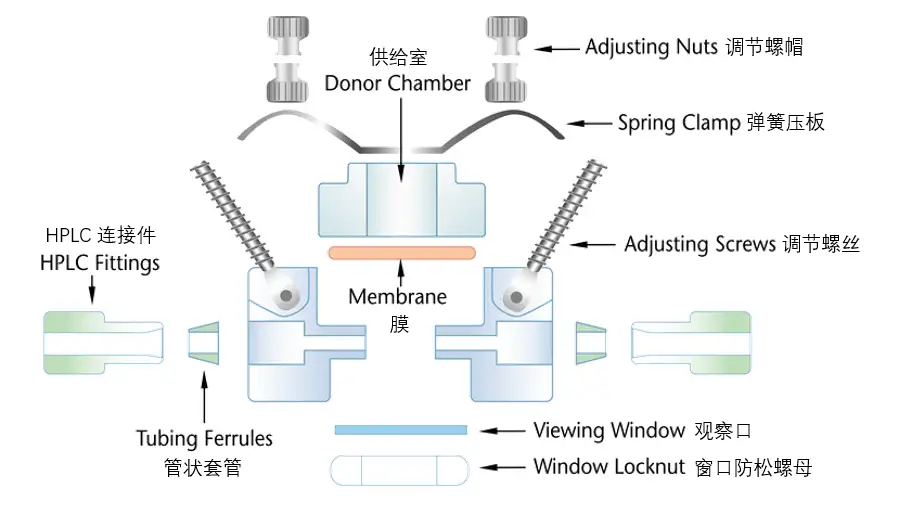
Fig.4 Schematic Diagram Detailing Individual Parts of a Horizontal Flow-through Cell图4 流通池各组成部分的水平示意图(拆卸后)
3 研究设计
3.1 皮肤
3.1.1 皮肤的选择
EMA在2018年发布的《Draft guideline on quality and equivalence of topical products》中仅推荐使用离体人类皮肤。PMDA规定可以使用离体大鼠、小鼠或猪的皮肤进行试验[8]。
在FDA 阿昔洛韦指南和IVPT指南中,均推荐采用离体人类皮肤[2, 14];但在USP <1724>[9]中规定,除采用离体人类皮肤外,因猪的皮肤与人类皮肤具有相对相似的形态,可作为研究的次要选择;然而由于啮齿类动物(如鼠、豪猪)的皮肤在形态学和角质层结构和厚度方面与人类皮肤显著不同,不鼓励使用;合成膜(包括模拟生物膜开发的合成膜)也不适用于IVPT方法,因为采用合成膜得到的结果不能反应透过皮肤的药物量和渗透速率,故用作临床预测的意义不大[17, 18]。
在外用药物制剂研究中,目前最常用的动物模型包括小鼠、大鼠、家兔和猪。其中,小鼠皮肤的角质层薄,通透性远大于人皮,但因其来源丰富、处理方便且结构与人皮相似,多在早期处方筛选中应用[19, 20]。家兔皮肤因其厚度约为人皮的一半,皮肤渗透性较大,难以作为经皮吸收预测模型,但由于家兔皮肤对药物吸收快的性质,常被用来考察药物的皮肤毒性[21, 22];此外,对于眼用半固体制剂,推荐采用家兔的角膜进行IVPT研究[23]。另有研究表明可用蛇皮代替猪耳皮肤和鼠皮进行IVPT研究,但如采用蛇皮,应注意控制皮肤的水合状态[24]。
猪由于在解剖学、生理学和代谢等方面与人类相似,被广泛应用于生物医学领域。大量文献表明,猪皮肤的组成、渗透性及体内代谢与人体较为相似[25]。因此,在外用制剂研究过程中,推荐采用猪的皮肤进行IVPT研究,常用的品种有巴马香猪、Yucatan小型猪、Landrace猪、Gottingen小型猪及Yorkshire猪等。
3.1.2 皮肤的纳入标准
FDA要求,对于离体人类皮肤,至少是18周岁的健康、正常、具有屏障完整性的雄性和/或雌性供体;所用的所有皮肤应有一致的来源(如年龄、种族),且为相同的解剖区域(如背部躯干;也可以使用其他生物膜,如直肠、阴道或角膜上皮组织,这取决于所评估药品的预期给药途径);同时,需规定每组供体皮肤的储存温度和时间,以及冻融循环次数;此外,FDA建议皮肤的厚度在(500±250)μm [2]。但USP要求,厚度通常在250~500 µm[9]。
猪皮一般选择1~3月龄的小型猪的腹部或背部皮肤,研究方案应包含猪的种族、年龄、皮肤厚度和解剖的区域等[26]。
朱慧勇等[27]研究表明将皮肤的新鲜度(将裸鼠皮肤于-20 ℃放置6个月,其皮肤屏障性能无明显差异)、厚度(相对标准偏差小于15%)、质量(相对标准偏差小于15%)、经皮水分散失(TEWL)达到稳态等因素作为皮肤模型的质量控制手段,可进一步增加研究结果的可靠性和准确度。
MORIN M等[28]研究表明将猪耳皮肤在不同的温度条件下放置不同的时间,新鲜皮肤相对其它条件具有较高的电阻且对亲水性化合物阿昔洛韦和疏水性水杨酸甲酯的渗透性均较低;在不同温度条件下放置不同时间的皮肤对阿昔洛韦在皮肤中的组织分布有显著影响;但根据水杨酸甲酯的研究结果可知,在不同的放置条件下,皮肤中均保存一定的酯酶活性。
根据上述指南及文献可知,在IVPT研究中对皮肤的控制至关重要,控制项目包括但不限于来源(年龄、种族)、解剖区域、新鲜度(放置时间、放置条件、冻融循环次数)、厚度、质量、稳态平衡和皮肤屏障完整性等。
3.1.3 皮肤的排除标准
FDA和EMA均要求,患有已知皮肤疾病(病史)的供体皮肤;带有纹身、妊娠纹或任何异常迹象的皮肤;表现出明显浓密的末端毛发的皮肤等应排除在外[2, 7]。
USP不推荐使用人或猪的全层皮肤,因其可能会人为地延迟药物的渗透,对实验造成挑战[9]。
3.1.4 皮肤的处理和保存
可以使用手术刀将特定解剖区域皮肤整块切下来,也可采用电动取皮机设置一定的厚度进行取皮。然后使用纯水或生理盐水冲洗皮肤外表面,并用镊子和刀片等小心刮去残留的皮下脂肪和血管等其他组织,然后反复冲洗干净,低温保存(建议储存于-20 ℃或-80 ℃冰箱中)[26]。此外,根据MORIN M等[28]的研究,所用皮肤最好在相同的保存条件(如-20 ℃或-80 ℃)下放置相同的时间,以避免不同的放置条件或时间造成药物组织分布的差异。
虽然可以采用温和的清洗或冲洗方式处理皮肤表面,但是,如果将皮肤浸泡在水溶液中几分钟,就有可能破坏皮肤屏障,应避免该处理方式。此外,也不建议采用刀片剃须、研磨抛光、胶带剥离,或采用乙醇或其它强极性溶剂处理皮肤,因为这些处理方式可能破坏皮肤屏障。含有显著的药物背景水平或含有的其他化合物可能干扰接收液中药物定量测定的皮肤供体,应排除在研究之外[2]。
因此,在上样前皮肤的解冻过程中,应尽可能的避免采用接收介质浸泡的方式,以免破坏皮肤的屏障完整性;实际操作中,通常可将皮肤在室温条件下放置约30 min即可。
3.1.5 皮肤的屏障完整性
FDA、EMA与PMDA均要求在试验前需进行皮肤屏障完整性测试[2, 7, 8]。FDA及USP均未明确说明试验后是否需要进行皮肤屏障完整性测试;但EMA要求,如果试验时间大于24 h,试验后需评估皮肤屏障完整性。可接受的屏障完整性测试方法包括TEWL、氚化水渗透、或电阻/电导值法。TEWL通常是首选方法[7, 9],不同于其他方法,TEWL是在不受外部空气影响的条件下,测定水分通过皮肤屏障的量,相对快速和方便。
FDA建议,对于人体躯干或大腿皮肤而言,采用TEWL屏障完整性测试法,合理的接受标准是每小时每平方米不大于约15 g水(即,≤15 g×m-2×h-1);但TEWL测量值,可能会因测试设备、操作方式和环境条件的不同而变化,如果通过实验数据表明,所选的接受标准可以适当地区分屏障完整性受损与合格的皮肤切片,也可以采用更高的接受标准(如,≤20 g×m-2×h-1)。采用氚化水屏障完整性测试法,将氚化水上样到皮肤表面5 min后移除,在移除30 min后取样,合理的皮肤屏障完整性接受标准是每平方厘米约1.5当量微升的氚化水(即:1.5 eq.μL×cm-2 or ~1.5 eq.mg×cm-2)。电阻/电导值法以电阻、电导或相关的电概念报告测试结果,用于表征流过皮肤的电流量,测试结果可以用电阻的倒数“电导”为单位进行描述;通常按以下步骤进行测定:①将皮肤安装到扩散池中,使角质层朝向供给室,皮肤下侧与接收介质接触,然后将皮肤表面温度平衡至(32±1)℃;②将可覆盖皮肤切片表面的少量离子溶液短暂的施加到皮肤角质层上;③将经皮电阻测定仪的一端电极与接收介质接触,另一端与供给室中离子溶液接触进行测定;④测定后将供给室中的离子溶液移出,用吸收性低脂棉将皮肤表面轻轻吸干,在上样前将皮肤与上面的干燥空气接触,并平衡足够长的时间,使角质层恢复到正常的水合状态[2]。
但应注意,测定方法不应不可逆的改变皮肤完整性。可以接受将水溶液上样到皮肤表面后,短暂的改变角质层水合状态的情况,但在上样前,经过足够长的时间后,角质层应可以恢复到正常的水合状态。未通过皮肤完整性测试的皮肤切片不应进行上样操作,但可作为无剂量空白对照皮肤切片[2]。
3.1.6 皮肤供体数和重复数
FDA要求在IVPT初步研究中,建议采用多个皮肤供体(如,4~6个皮肤供体),且每个试验组的每个供体至少4个重复皮肤切片[2];EMA要求每个试验组皮肤供体的数量不应少于 12 个,每个供体至少 2 个重复[7];PMDA要求每个试验组至少进行6次测定[8]。
通常,在人群中的不同个体之间,或同一个体不同解剖区域的皮肤渗透性,存在10倍的差异是很常见的,且在IVPT研究过程中涉及的试验参数较多,操作难度较大。在生物等效性评估中,建议按照FDA或EMA的皮肤供体和重复数要求,采用统计学方法对IVPT的一致性进行评估。PMDA要求的6次测定,适用于外用制剂上市后处方变更的评估,不适用于产品的上市申请;然而,在前期的处方开发阶段,可用于工艺参数的优化和评估。
3.2 IVPT方法开发
适用于支持外用制剂生物等效性论证的IVPT方法的开发,通常需要进行一系列系统的探索性研究,证明所选择的IVPT方法参数是合理性或必要的,相关的参数包括:设备、皮肤来源、皮肤类型、皮肤处理方法、皮肤屏障完整性测试方法及接受标准、上样量、上样方式、接收介质、剂量维持时间、研究持续时间、接受液取样时间点,以及样品分析方法的选择等。
3.2.1 上样量及上样方式
USP要求,在IVPT研究过程中,设备供给室应保存开放,采用有限剂量,建议配方制剂的上样量在2~15 mg×cm-2之间(方法开发期间在5~10 mg×cm-2之间)。FDA推荐采用相同皮肤供体的重复皮肤切片进行不同剂量的平行对比研究,优化所需的上样量,需要考虑的因素包括:(1)采用相同的上样方式;(2)通量曲线的重现性;(3)上样量和剂量维持时间对通量曲线的影响;和(4)接收液中不同取样时间点的药品浓度大致范围(与样品分析方法的定量限度相关)[9]。EMA要求,除非产品特性概要中另有规定,设备供给室应采用开放式,配方制剂的上样量在2~15 mg×cm-2之间,并需对上样方式进行确认,确保可将配方制剂均匀一致的涂抹到皮肤上,且上样量误差应在±5%之间[7]。
上样时根据制剂的特性可以选择按重量和按体积2 种方式进行。黏度高、流动性差的制剂可按重量方式上样;黏度低、流动性好的制剂可按体积方式上样[29]。USP建议使用外置活塞式移液器,以确保上样的精度;对于半固体剂型,如软膏剂、乳膏剂和某些不像溶液一样可以流动的凝胶剂,可以使用玻璃棒或玻璃瓶的底部将制剂均匀地涂覆到上样区域[9]。FDA进一步说明,在按体积上样后,需采用高效液相色谱玻璃瓶的平底、玻璃棒或毛细管的圆端,将分配到皮肤上的药品涂覆均匀[2]。
由上述要求可知,在IVPT研究中,为模拟药物临床使用环境,除非产品特性概要或说明书中另有规定,供给室应采用开放式,并将配方制剂的上样量控制在2~15 mg×cm-2之间;同时,需根据外用制剂的不同特性,摸索确定相应的上样方式,确保上样误差控制在±5%之间,以便于IVPT方法开发过程中质量平衡的评估。
3.2.2 接收介质
IVPT研究中所用接收介质的组成和pH应根据其与皮肤的兼容性以及药物在接收介质中的稳定性和溶解度进行确认。EMA要求,应采用水性缓冲液作为接收介质,在整个试验期间,接收介质中活性物质的最大浓度不应超过在该接收介质中活性物质最大溶解度的30%[7]。PMDA要求接收介质应不影响皮肤的屏障完整性,可采用水或缓冲液,如需要可添加表面活性剂,如吐温80[≤1%(w/v)]、聚乙二醇400[≤40%(w/v)],但不能添加有机溶剂[8]。
FDA要求,药物在接收介质中的溶解度,应经过三次重复检测经验确定,确保其超过IVPT正式研究中最高样品浓度,理想情况下是一个数量级。对于疏水性药物,推荐在基于生理缓冲盐的接收介质中添加0.1%(w/v)聚氧乙烯20油醚(CAS:9004-98-2)以提高溶解度;如有必要,可在接收介质中略微增加聚氧乙烯20油醚的浓度[例如,从0.1%(w/v)到0.2%(w/v)],通常足以满足大多数疏水药物溶解度的要求,但不应超过6%;其他改善药物在接收介质中溶解度的策略可能会改变皮肤的渗透性(例如,在接收介质中加入有机溶剂和醇),使IVPT方法失效,不建议使用;此外,无论研究时间的长短,建议在接收介质中加入一种抗微生物剂(例如,0.1%叠氮化钠或0.01%硫酸庆大霉素),以减轻在整个研究期间扩散池中细菌对真皮和/或表皮的潜在分解,如果采用其它抗微生物剂,应阐述选择的理由[2]。
因此,对于一个优化良好的接收介质应满足以下要求:①一般选择生理盐水或pH 6.8~7.4的磷酸盐缓冲溶液[26](离子强度约为0.15)作为接收介质,不能添加有机溶剂或醇,以免破坏皮肤屏障完整性;对于疏水性药物,可添加对皮肤影响较小的表面活性剂,如吐温80[≤1%(w/v)]、聚乙二醇400[≤40%(w/v)]、聚氧乙烯20油醚[≤6%(w/v),通常为0.1%(w/v)];②无论研究时间的长短,建议在接收介质中加入一种抗微生物剂(例如,0.1%(w/v)叠氮化钠或0.01%(w/v)硫酸庆大霉素),以减轻研究期间细菌对真皮和/或表皮的潜在分解;③接收介质对药物的溶解度应符合漏槽条件,即接收介质的体积为药物饱和溶液所需介质体积的3~10倍;④在整个试验期间,药物在接收介质中稳定;⑤为避免在试验期间产生气泡,通常需对接收介质进行脱气处理。
3.2.3 皮肤温度
FDA和EMA均要求,在整个研究过程中,每个皮肤切片表面的温度应保持在(32±1)℃[2,7],PMDA要求保持在(32±0.5)℃。对于内用产品(例如,直肠和阴道用品),USP要求温度为(37±1)℃[9]。
3.2.4 研究持续时间及取样时间点
所选择的取样时间点和研究持续时间应足以表征药物的皮肤药代动力学,理想情况下应包括一个足够完整的通量分布,以识别最大(峰值)通量和随后的通量下降情况。在研究期间,如果药品持续保留在皮肤上面,其可能会持续递送药物,在最大(峰值)通量之后时间点可能不会通量下降的现象。此时,在IVPT方法开发时,可在上样一段时间后将样品擦去,然后持续监控接收液一定时间,使之呈现出通量下降的情况。所选的取样频率应足以为通量曲线提供合适的分辨率,FDA要求在整个研究期间,推荐至少采用8个非零取样点[2]。
USP建议研究持续时长在12~72 h之间,一般应大于等于24 h[9];EMA建议研究持续时长以24 h为宜,如果大于24 h,在试验结果后应对皮肤的屏障完整性进行评估[7];PMDA要求最长时间不得过24 h [8]。
各国对IVPT研究持续时间存在一定差异,但通常建议将其控制在24 h内,以避免因研究时间过长,导致皮肤屏障完整性破坏。
3.2.5 药物组织分布与质量平衡
EMA要求活性物质渗透进入接收液的总量、皮肤组织中活性物质的总量和残留在皮肤或试验设备上的总量之和应在90%~110%范围内[7];根据研究的目的,可能需要提供关于药物组织分布(角质层、活性表皮层和真皮层中药物的量)的评估。
对于药物组织分布的评估,FDA建议在实验结束时,可先用棉签去除皮肤表面多余的制剂,然后用胶带剥离法去除角质层,再将皮肤切片放置在铝箔上,于60 ℃干热条件下孵育2~3 min,最后用镊子将表皮层与真皮层分离开(通过刮擦)[9]。
3.3 样品分析方法的选择
在IVPT方法开发过程,样品分析方法的选择至关重要,如残留在皮肤上和皮肤组织中样品的处理、渗透进入接收液总量的分析方法选择等。
3.3.1 残留在皮肤上的样品
建议在实验结束时先用棉签去除皮肤表面大部分的配方制剂,然后用胶带剥离。在胶带剥离时,EMA要求胶带应满足以下要求:①与皮肤表面磨擦后,不损失重量;②药物易于从粘附在胶带上的角质层中提取;③胶带的粘合剂或其他成分不干扰药物的定量测定;④胶带应有足够的粘合力,即使采用较少的胶带数量也可以除去大部分的角质层。在方法开发的过程中,所用的胶带剥离程序必须确保大部分角质层(≥75%)被采样,可以根据TEWL接受标准(或其它相关技术)确定所需胶带的最少和最大数量[7]。
3.3.2 皮肤组织中药物滞留量
成铃等[30]将皮肤剪碎后,在45 ℃条件下,用体积分数为50%的甲醇超声提取20 min,盐酸特比萘芬含量在(12.50~20.21)μg×cm-2。陈积等[31]将皮肤剪碎后,用甲醇涡旋5 min、超声60 min提取吉马酮和维A酸,含量分别为(0.7792±0.1424)μg×cm-2和(1.4895±0.2848)μg×cm-2。宋娟等[32]将皮肤剪碎后,用甲醇超声5 min提取活性成分。陈观凤等[33]将皮肤剪碎后,用无水乙醇超声处理30 min,黄酮素脂质体中黄藤素含量为(13.02±1.05)μg×cm-2。吴昭全等[34]将皮肤剪碎后,用甲醇浸泡12 h提取羟基积雪草酸,结果在(0.2963~1.3656)μg×cm-2。
Cross, S.E[35]等将皮肤剪碎后,采用水-乙腈-磷酸(760:233:7)涡旋超声30 s对水杨酸甲酯、水杨酸乙二醇酯和水杨酸三乙醇胺酯进行提取。Jacques-Jamin C[36]等采用Soluene-350加热至60 ℃对皮肤组织进行消解后,对肉桂酸、苯甲酸和6-甲基香豆素进行测定。JANKOVSKAJA S[37]等采用pH为12.0的131 mmol×L-1氯化钠对皮肤中的色氨酸和犬尿氨酸进行超声提取。
一般情况下,皮肤组织内样品可采用高比例有机溶剂(如甲醇、乙醇和乙腈等)或强碱性溶液等进行超声、振荡或浸泡提取,亦可以采用消解试剂(如Soluene-350)将皮肤组织消解后进行测定;通常在方法开发过程中,需对不同溶剂、不同提取方式进行筛选,选择提取时间短、操作方便且能保持药物稳定的提取方式。此外,由于皮肤组织的复杂性及药物滞留量较低(通常为μg×cm-2级别),在色谱方法选择时,需重点关注方法的检测灵敏度及皮肤组织对样品检测的干扰。因透皮试验研究中所用皮肤厚度通常为(500±250)μm,对于皮肤组织提取前是否需要剪碎处理,可通过剪碎与整块皮肤切片的提取结果对比确定,推荐操作步骤为:先对整块皮肤切片进行提取,然后将初次提取后的皮肤切片剪碎后再进行提取,如果后者的提取结果远小于整块皮肤切片的提取结果(如小于5%),则无需进行剪碎处理。
3.3.3 接收液中的样品
USP<1724>指出,接收液中药物的浓度一般在pg~ng范围内[9];郭涤亮[38]等报道阿昔洛韦乳膏欧美不同国家参比制剂单位面积药物渗透速率约在10~60 ng×cm-2×h-1之间。
在对接收液中样品进行测定时,通常采用高效液相色谱-质谱法或超高效液相色谱-质谱法。FDA要求采用多点校准曲线(6~8个)进行测定,同时应符合FDA生物分析方法验证行业指南要求[2, 39]。此外,在方法开发过程中,因样品量较低,需对样品处理方法进行考察,如滤膜过滤或离心等,如采用滤膜过滤进行处理,需确保滤膜对样品无吸附。
3.4 数据统计分析
通常,IVPT采用药物单位面积的最大渗透速率(Jmax,单位为ng×cm-2×h-1)和总累积渗透量(Atotal,单位为ng×cm-2)来表示。FDA、EMA和PMDA在数据统计和等效性判定依据方面存在差异,下面分别对其进行介绍。在实际研究工作中,可根据药物不同的研究阶段、所用供体数量和重复数,采用合适的方法对结果进行分析。
Jmax和Atotal的统计分析过程和接受标准一致,下文仅以Jmax为例进行介绍。
3.4.1 FDA
在统计分析过程中,要求受试制剂和对照制剂各至少4个供体组,每组供体至少3个可用的重复皮肤切片,推荐采用SAS软件进行统计分析,其统计分析过程和可接受标准,概况如下:①计算对照制剂各组供体间Jmax的偏差( );②如果≥0.294,采用标度的平均生物等效性(scaled average bioequivalence,简称SABE)评估法,否则采用双侧T检验(TOST)的平均生物等效性(average bioequivalence,简称ABE)评估法;③对于SABE评估法,如果的95%置信区间小于等于0,且对照制剂和受试制剂几何平均数的比率在0.8000~1.2500之间,可判定为等效;对于ABE评估法,如果的90%置信区间在0.8000~1.2500之间,可判定为等效。
);②如果≥0.294,采用标度的平均生物等效性(scaled average bioequivalence,简称SABE)评估法,否则采用双侧T检验(TOST)的平均生物等效性(average bioequivalence,简称ABE)评估法;③对于SABE评估法,如果的95%置信区间小于等于0,且对照制剂和受试制剂几何平均数的比率在0.8000~1.2500之间,可判定为等效;对于ABE评估法,如果的90%置信区间在0.8000~1.2500之间,可判定为等效。
3.4.2 EMA
受试制剂与对照制剂均值比率的90%置信区间应在80.00%~125.00%之间;对于小规格和扩散量有限的药物产品,如果观察到具有高变异性,可通过临床研究,放宽90%置信区间,采用更宽松的接受标准69.84%~143.19%。Jmax常呈非正态分布,在计算过程中需进行对数转换,具体计算过程如下。
将获得的受试制剂(T)和对照制剂(R)各24个数据,计算受试制剂组和对照制剂组各个Jmax的相互比值T/R,得到576个T/R值,然后对这些T/R值进行对数转换。使用下式计算置信区间:
其中,X为所有T/R值对数转换后的平均值,n为样品量,t为n-1个自由度(t)的90%置信区间的t值(n=6,t=2.015;n=12,t=1.796;n=18,t=1.740;n=24,t=1.714),s为所有T/R值对数转换的标准偏差。将所得结果取反对数,即为置信区间。
3.4.3 PMDA
在规定的试验时间或渗透速率达到平台期后的1个时间点,以及和该时间点对应的渗透速率的一半所对应的时间点,受试制剂与标准制剂的平均渗透速率之比均应在 0.7~1.3 之间。此外,PMDA要求受试制剂渗透速率的偏差与标准制剂相比,应相同(可采用F检验进行判断)或更小。
4 结语
本文汇总了CDE、FDA、EMA和PMDA等国家关于IVPT研究的政策法规,目前国内陆续发布了外用制剂相关研究技术指南,关于外用制剂中经皮研究的文献也越来越多,但尚未有统一的标准进行规定,包括体外透皮试验的数据统计分析等,各国要求也不尽相同。外用制剂本身具有工艺复杂性,在IVPT研究中,亦有很多技术难点,如皮肤的选择、皮肤的纳入标准、样品的上样方式、接收介质的选择、药物组织分布研究和样品分析方法考量等。伴随着我国对外用制剂研究的越加深入,希望结合上述对IVPT研究技术参数的讨论,重点参照2022年USP<1724>和FDA IVPT指南,结合实际需要,尽早统一IVPT研究技术要求。
参考文献
[1] Center for Drug Evaluation. Guiding principles of research on generic drugs for external use of skin chemicals (Trial) [S]. 2021.[2] FDA. Guidance for industry in vitro permeation test studies for topical drug products submitted in ANDAs [S]. 2022.[3] FDA. Physicochemical and structural (Q3) characterization of topical drug products submitted in ANDAs [S]. 2022.[4] USP-NF 2023. General Chapters 3: Topical and transdermal drug products-product quality tests[M]. 2023.[5] LU M, XING H, CHEN X, et al. Advance in bioequivalence assessment of topical dermatological products [J]. Asian J Pharm Sci, 2016, 11(6): 700-707.[6] SHAH V P, YACOBI A, RĂDULESCU F S, et al.A science based approach to topical drug classification system (TCS) [J]. Int J Pharm, 2015, 491(1/2): 21-25.[7] EMA, CHMP: draft guideline on quality and equivalence of topical products [S]. 2018.[8] PMDA. Guideline on biological equivalence for formulation changes of topical products (semisolid products and patches). [S]. 2010. [9] PF48(3). General Chapters 1724: Semisolid drug products-performance tests[S]. 2022.[10] Center for Drug Evaluation. Technical evaluation requirements for the new registration classification of generic drugs for external use on skin (Draft for solicitation of comments) [S]. 2018.[11] Center for Drug Evaluation. Guiding principles for pharmaceutical research of transdermal patches of chemical generic drugs (Trial) [S]. 2020.[12] Center for Drug Evaluation. Guiding principles for clinical trial of locally applied locally acting drug [S]. 2022.[13] FDA. SUPAC-SS: nonsterile semisolid dosage forms; scale-up and post-approval changes: chemistry, manufacturing and controls; in vitro release testing and in vivo bioequivalence documentation [S]. 1997.[14] FDA. Product-specific guidances for generic drug development. Graft guidance on Acyclovir [S]. 2016.[15] EMA, CHMP: guideline on quality of transdermal patches [S]. 2014.[16] PMDA. Guideline on biological equivalence for formulation changes of topical products (semisolid products and patches): questions and answers [S]. 2010.[17] SZYMAŃSKI Ł, JĘDERKA K, CIOS A, et al.A simple method for the production of human skin equivalent in 3D, multi-cell culture [J].Int J Mol Sci, 2020, 21(13): 4644.[18] OSHIMA S, SUZUKI C, YAJIMA R, et al.The use of an artificial skin model to study transdermal absorption of drugs in inflamed skin [J].Biol Pharm Bull, 2012, 35(2): 203-209.[19] ROBERTS M E, MUELLER K R.Comparisons of in vitro nitroglycerin (TNG) flux across yucatan pig, hairless mouse, and human skins [J].Pharm Res, 1990, 7(6): 673-676.[20] SATO K, SUGIBAYASHI K, MORIMOTO Y.Species differences in percutaneous absorption of nicorandil [J].J Pharm Sci, 1991, 80(2): 104-107.[21] NICOLI S, PADULA C, AVERSA V, et al.Characterization of rabbit ear skin as a skin model for in vitro transdermal permeation experiments: histology, lipid composition and permeability [J].Skin Pharmacol Physiol, 2008, 21(4): 218-226. [22] SASAKI H, KOJIMA M, NAKAMURA J, et al.Acute toxicity and skin irritation of pyrrolidone derivatives as transdermal penetration enhancer [J].Chem Pharm Bull (Tokyo), 1990, 38(8): 2308-2310.[23] AL-GHABEISH M, Xu X M, KRISHNAIAH Y S, et al. Influence of drug loading and type of ointment base on the in vitro performance of acyclovir ophthalmic ointment [J]. Int J Pharm, 2015, 495(2): 783-791.[24] PRAÇA F S G, MEDINA W S G, ELOY J O, et al. Evaluation of critical parameters for in vitro skin permeation and penetration studies using animal skin models [J]. Eur J Pharm Sci, 2018, 111: 121-132.[25] SIMON G A, MAIBACH H I.The pig as an experimental animal model of percutaneous permeation in man: qualitative and quantitative observations--an overview [J].Skin Pharmacol Appl Skin Physiol, 2000, 13(5): 229-234.[26] The Transdermal Drug Administration Committee of the World Federation of Chinese Medicine Societies. Discussion of professional consensus on the technical practice of the in vitro permeation test for topical and transdermal preparations[J]. Chinese Journal of Modern Applied Pharmacy(中国现代应用药学), 2022, 38(8): 1128-1132.[27] ZHU H Y, WU Y B, LU W D, et al. Skin models and critical quality control for the studies of transdermal drug delivery systems [J]. Chinese Journal of Pharmaceuticals(中国医药工业杂志), 2022, 53(5): 592-600.[28] MORIN M, RUNNSJö A, RUZGAS T, et al.Effects of storage conditions on permeability and electrical impedance properties of the skin barrier[J]. Int J Pharm, 2023.122891.[29] MA H, ZHAO J, GE Q H, et al. Overview and application examples of in vitro permeation test (IVPT) for dermatological products [J]. Chinese Journal of Pharmaceuticals(中国医药工业杂志), 2021, 52(8): 1010-1018.[30] CHENG L, WANG B T. Percutaneous absorption and content determination of terbinafine hydrochloride cream [J]. Chinese Journal of Hospital Pharmacy(中国医院药学杂志), 2015, 35(6): 518-522.[31] CHEN J, ZHAO X Q, MA Y Q, et al. Preparation and in vitro transdermal permeation of zedoary turmeric oil compound liposomes gel [J]. Chinese Journal of New Drugs(中国新药杂志), 2018, 27(7): 830-838. [32] SONG J, LV C Z, WANG D, et al. Study on the preparation and in vitro transdermal penetration of epigallocatechin-3-gallate binary ethosomes [J]. Journal of Practical Dermatology(实用皮肤病学杂志), 2020, 13(6): 329-332.[33] CHEN G F, YANG F Z, ZHENG Z J, et al. Preparation and in vitro transdermal properties of palmatine liposomes [J]. Traditional Chinese Drug Research & Clinical Pharmacology(中药新药与临床药理), 2022, 31(1): 115-119.[34] WU S Q, JIANG Y L, LUO H Q, et al. Preparation and transdermal permeability in vitro and in vivo of madecassic acid solid lipid nanoparticles gel [J]. China Pharmacist(中国药师), 2022, 25(7): 1150-1156.[35] CROSS S E, ANDERSON C, ROBERTS M S. Topical penetration of commercial salicylate esters and salts using human isolated skin and clinical microdialysis studies [J]. Br J Clin Pharmacol, 1998, 46: 29-35.[36] JACQUES-JAMIN C, DUPLAN H, ROTHE H, et al. Comparison of the skin penetration of 3 metabolically stable chemicals using fresh and frozen human skin. Skin Pharmacol. Physiol [J]. 2017, 30: 234–245.[37] JANKOVSKAJA S, ENGBLOM J, REZELI M, et al. Non-invasive skin sampling of tryptophan/kynurenine ratio in vitro towards a skin cancer biomarker[J]. Sci. Rep. 2021, 11: 678.[38] GUO D L, WANG Y M, MA Y N. Considerations on the selection of reference-listed drugs for the research and evaluation of topical dermatologic generic drugs [J]. Chinese Journal of New Drugs(中国新药杂志), 2019, 28(6): 661-665.[39] FDA. Bioanalytical Method Validation Guidance for Industry [S]
